 RESEARCH
RESEARCH| Home · Teaching · Personal (in Spanish only) |
Juan J. Meléndez
|
RESEARCH
1. Mechanisms of plastic deformation of ceramic polycrystals at high temperature
 Advanced ceramics have been preferential systems for scientists
and engineers throughout the 20th century. This interest has been
greatly based upon the particular physical properties exhibited by
some of these systems, which make them optimum for certain
functional applications; for instance, UO2 is used in nuclear
reactors, ZrO2 is a component of fuel cells and some YBaCuO are
superconductors. Some other ceramics are interesting due to their
potential structural applications; for instance, the
superplasticity of ZrO2 (similar to that of the metals) was
discovered in 1986. Anyway, the study of the mechanical behavior
of ceramics is relevant; in the case of functional materials,
because their integrity must be guaranteed under working
conditions. In structural ones, the mechanical properties are
inherently interesting.
Advanced ceramics have been preferential systems for scientists
and engineers throughout the 20th century. This interest has been
greatly based upon the particular physical properties exhibited by
some of these systems, which make them optimum for certain
functional applications; for instance, UO2 is used in nuclear
reactors, ZrO2 is a component of fuel cells and some YBaCuO are
superconductors. Some other ceramics are interesting due to their
potential structural applications; for instance, the
superplasticity of ZrO2 (similar to that of the metals) was
discovered in 1986. Anyway, the study of the mechanical behavior
of ceramics is relevant; in the case of functional materials,
because their integrity must be guaranteed under working
conditions. In structural ones, the mechanical properties are
inherently interesting.
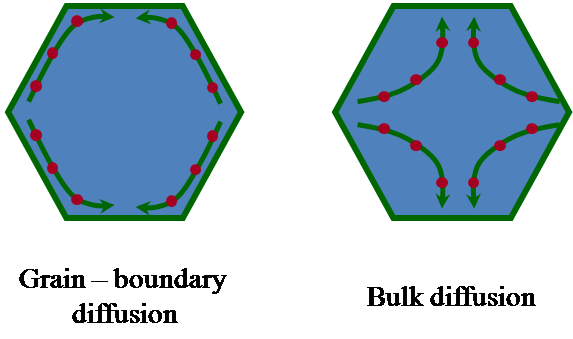 In general terms, creep denotes the process of plastic deformation
of a material at temperatures above 0.5Tm, where Tm is the melting
temperature of the material, under the action of an external, an
generally uniaxial, stress. The relevant physical properties in a
creep process are the strain rate, the strain to failure and the
creep stress, among others. In polycrystals, the plastic
deformation proceeds either by diffusion or by grain boundary
sliding; the latter does not involve any change in the grain
shapes. In all cases, internal stresses are generated at the
grains during the plastic deformation, which must be released in
order to avoid the eventual failure of the material. The process
by which the internal stresses are relaxed is called accommodation
mechanism, and uses to be diffusional in nature. The various
mechanisms differ to each other by the process responsible of the
deformation (diffusion or grain boundary sliding) and by the
accommodation process (which determines the strain rate at which
the material creeps).
In general terms, creep denotes the process of plastic deformation
of a material at temperatures above 0.5Tm, where Tm is the melting
temperature of the material, under the action of an external, an
generally uniaxial, stress. The relevant physical properties in a
creep process are the strain rate, the strain to failure and the
creep stress, among others. In polycrystals, the plastic
deformation proceeds either by diffusion or by grain boundary
sliding; the latter does not involve any change in the grain
shapes. In all cases, internal stresses are generated at the
grains during the plastic deformation, which must be released in
order to avoid the eventual failure of the material. The process
by which the internal stresses are relaxed is called accommodation
mechanism, and uses to be diffusional in nature. The various
mechanisms differ to each other by the process responsible of the
deformation (diffusion or grain boundary sliding) and by the
accommodation process (which determines the strain rate at which
the material creeps).
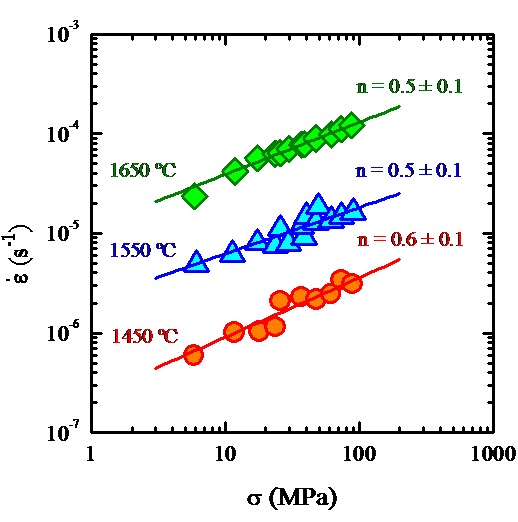
 The identification of the deformation mechanisms in polycrystals
takes place by a sequence of mechanical tests, which allow
characterize the process macroscopically, and microstructural
observations of the samples before and after the deformation. The
comparison of both microstructures allows one obtain information
about the primary and accommodation processes and thence to
identify the deformation mechanism. Since 1998 I have been
studying the plastic deformation at high temperatures of ceramic
materials in collaboration with the University of Sevilla. These
ceramics have been either oxidic (SiC whiskers-reinforced alumina
[1, 4], aluminum titanate stabilized with oxides and doped with
mullite [2], zircon [3] and mullite [10]) and, mainly, non-oxidic
(like ZrB2 [8], SiC [21] and, above all, Si3N4 [7, 13, 14]). Among
these works, the study of two gas-pressure sintered silicon
nitride ceramics containing a secondary glassy phase is
outstanding. The high temperature mechanical tests (performed at
temperatures between 1450 ºC and 1750 ºC) yielded a stress
exponent systematically lower than one (with an average value of
about 0.5) for both samples under all the experimental conditions.
The microstructural observations were consistent, in principle,
with a grain boundary mechanism accommodated by
solution-precipitation. However, the results were surprising
because all the commonly invoked models for creep in this type of
materials predicts stress exponents equal to 1 or between 1 and 2.
The identification of the deformation mechanisms in polycrystals
takes place by a sequence of mechanical tests, which allow
characterize the process macroscopically, and microstructural
observations of the samples before and after the deformation. The
comparison of both microstructures allows one obtain information
about the primary and accommodation processes and thence to
identify the deformation mechanism. Since 1998 I have been
studying the plastic deformation at high temperatures of ceramic
materials in collaboration with the University of Sevilla. These
ceramics have been either oxidic (SiC whiskers-reinforced alumina
[1, 4], aluminum titanate stabilized with oxides and doped with
mullite [2], zircon [3] and mullite [10]) and, mainly, non-oxidic
(like ZrB2 [8], SiC [21] and, above all, Si3N4 [7, 13, 14]). Among
these works, the study of two gas-pressure sintered silicon
nitride ceramics containing a secondary glassy phase is
outstanding. The high temperature mechanical tests (performed at
temperatures between 1450 ºC and 1750 ºC) yielded a stress
exponent systematically lower than one (with an average value of
about 0.5) for both samples under all the experimental conditions.
The microstructural observations were consistent, in principle,
with a grain boundary mechanism accommodated by
solution-precipitation. However, the results were surprising
because all the commonly invoked models for creep in this type of
materials predicts stress exponents equal to 1 or between 1 and 2.
.jpg) For instance,
this is so in the Wakai "step" model for solution - precipitation.
Essentially, it is assumed that the relaxation of the stresses
generated during grain boundary sliding takes place by a
sequential process consisting in solution of crystalline material
in the secondary glassy phase followed by diffusion along this
phase and finally precipitation at stress-free grain boundaries.
The slowest of these mechanism is the rate-controlling one. Thus,
if the lowest one is transport along the secondary phase, the
mechanism is said to be diffusion-controlled, and is characterized
by a stress exponent n = 1. On the contrary, if the slowest
processes are those of solution or precipitation, the mechanism is
said to be interface reaction - controlled, and is consistent with
stress exponent n = 1 or n = 2. The traditional mechanism of
solucion-precipitation is not consistent with stress exponent
lower than one in any case.
For instance,
this is so in the Wakai "step" model for solution - precipitation.
Essentially, it is assumed that the relaxation of the stresses
generated during grain boundary sliding takes place by a
sequential process consisting in solution of crystalline material
in the secondary glassy phase followed by diffusion along this
phase and finally precipitation at stress-free grain boundaries.
The slowest of these mechanism is the rate-controlling one. Thus,
if the lowest one is transport along the secondary phase, the
mechanism is said to be diffusion-controlled, and is characterized
by a stress exponent n = 1. On the contrary, if the slowest
processes are those of solution or precipitation, the mechanism is
said to be interface reaction - controlled, and is consistent with
stress exponent n = 1 or n = 2. The traditional mechanism of
solucion-precipitation is not consistent with stress exponent
lower than one in any case.
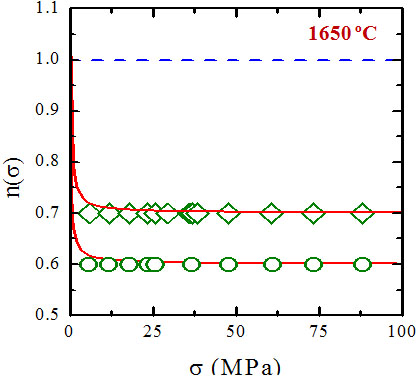 In the original "step" model, Wakai assumes that the solute
chemical potential within the grain is identically equal to the
one in the supersaturation conditions which give rise to the
solution. Using this fact, Wakai calculates a stress exponent
which is a function of the applied stress and the temperature; in
addition, the numerical values of this exponent are not only
greater than one, but also than one hundred in the typical stress
and temperature ranges for silicon nitride. This actually
constitutes a physical inconsistency of the model. The solution to
this inconsistency was to consider in greater detail the
solution-precipitation process [12]. In particular, there are two
terms which were not included by Wakai in his original
calculation; the first of them corresponds to the free energy
increment due to the different solute volumes in solution and in
solid phase. The second term is associated to the change in the
solute chemical potential within the grain and supersaturated at
the step under stress. If these two terms (particularly the
second) are included, the stress exponent not only acquires the
right order of magnitude, but also can be naturally lower than one
under certain conditions. Reference [12] thus contains the final
form of the solution-precipitation model.
In the original "step" model, Wakai assumes that the solute
chemical potential within the grain is identically equal to the
one in the supersaturation conditions which give rise to the
solution. Using this fact, Wakai calculates a stress exponent
which is a function of the applied stress and the temperature; in
addition, the numerical values of this exponent are not only
greater than one, but also than one hundred in the typical stress
and temperature ranges for silicon nitride. This actually
constitutes a physical inconsistency of the model. The solution to
this inconsistency was to consider in greater detail the
solution-precipitation process [12]. In particular, there are two
terms which were not included by Wakai in his original
calculation; the first of them corresponds to the free energy
increment due to the different solute volumes in solution and in
solid phase. The second term is associated to the change in the
solute chemical potential within the grain and supersaturated at
the step under stress. If these two terms (particularly the
second) are included, the stress exponent not only acquires the
right order of magnitude, but also can be naturally lower than one
under certain conditions. Reference [12] thus contains the final
form of the solution-precipitation model.
2. Resolution of crystalline structures by X-ray powder diffractometry
 The development the Organic and Inorganic Chemistries nowadays
gives rise to the synthesis of a great number of artificial
substances. The interest of these compounds lies on their multiple
applications: as pigments, catalyzers, agents with luminescent
activity and, above all, on their potential biological activity
(antitumoral, analgesic, antibiotic, etc.). That is the reason why
it is important to know the crystalline structure of some of them.
The development the Organic and Inorganic Chemistries nowadays
gives rise to the synthesis of a great number of artificial
substances. The interest of these compounds lies on their multiple
applications: as pigments, catalyzers, agents with luminescent
activity and, above all, on their potential biological activity
(antitumoral, analgesic, antibiotic, etc.). That is the reason why
it is important to know the crystalline structure of some of them.
In this context, an essential difficulty arises due to that, in
general, one does not have single crystals with appropriate size
and quality; instead, a good number of the artificial substances
are obtained directly as a powder. This fact makes the
conventional techniques of structural resolution from X-ray
diffractometry not to be valid, so that new resolution strategies
must be designed and optimized from powder diffraction diagrams.
These correspond to 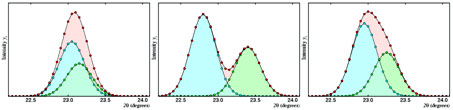 two-dimensional
representations of the diffracted intensity as a function of the
diffraction angle; since, in these cases, the Bragg angle can be
the same for different families of planes, the powder diffraction
diagrams are characterized because their maxima are generally
strongly overlapped. Actually, the difficulty associated to the
univocal separation of the different reflections merging at the
same maxima is one of the main limitations of the powder
diffractometry.
two-dimensional
representations of the diffracted intensity as a function of the
diffraction angle; since, in these cases, the Bragg angle can be
the same for different families of planes, the powder diffraction
diagrams are characterized because their maxima are generally
strongly overlapped. Actually, the difficulty associated to the
univocal separation of the different reflections merging at the
same maxima is one of the main limitations of the powder
diffractometry.
Formally, the structural resolution of a crystal consists on determine the relative positions of each atom in the asymmetric unit within the unit cell. It is a mathematical problem which can be solved if the structure factors corresponding to a number of hkl directions of the reciprocal space; these structural factors are complex quantities whose phases contain the information about the atomic positions. However, in a diffraction experiment one just gets information about the diffracted intensity for each reflection, which is proportional to the square of the corresponding structure factor; alternatively, one has information about the modulus of the structure factors, but neither about their phase and, therefore, nor in principle about the atomic positions. This fact is the well known "phase problem", where the main difficulty of the structural resolution lies.
 The phase problem is generally solved applying any of the
reciprocal space methods, such as the direct methods, whose
philosophy essentially consists of recognize that, somehow, the
information about the phases is diluted in the set of observed
structure factors moduli. Depending on the structure, a number of
phases can be arbitrarily assigned, and all the others are
obtained by the triplet rule. Direct methods allow solve the
structure of a crystal provided that one has at least ten
reflections (that is, distinguishable non-overlapped maxima) for
each atom in the asymmetric unit. When such a requirement is not
fulfilled (for instance, because the molecule is complex, because
the experimental data are not good enough, or because de
particular conditions make impossible the deconvolution of the
peaks) the so-called real space methods must be used. In this case
no phases are assigned to the structure factors (and thence the
phase problem is not directly solved). Instead, the starting point
is the knowledge of the connectivity of the isolated molecular
(which is generally known from spectroscopy), and the structural
resolution proceed by a Monte Carlo algorithm oriented to the
minimization of a certain cost function. Finally, the various
interesting parameters are refined by the Rietveld method to reach
a reasonable degree of precision [15].
The phase problem is generally solved applying any of the
reciprocal space methods, such as the direct methods, whose
philosophy essentially consists of recognize that, somehow, the
information about the phases is diluted in the set of observed
structure factors moduli. Depending on the structure, a number of
phases can be arbitrarily assigned, and all the others are
obtained by the triplet rule. Direct methods allow solve the
structure of a crystal provided that one has at least ten
reflections (that is, distinguishable non-overlapped maxima) for
each atom in the asymmetric unit. When such a requirement is not
fulfilled (for instance, because the molecule is complex, because
the experimental data are not good enough, or because de
particular conditions make impossible the deconvolution of the
peaks) the so-called real space methods must be used. In this case
no phases are assigned to the structure factors (and thence the
phase problem is not directly solved). Instead, the starting point
is the knowledge of the connectivity of the isolated molecular
(which is generally known from spectroscopy), and the structural
resolution proceed by a Monte Carlo algorithm oriented to the
minimization of a certain cost function. Finally, the various
interesting parameters are refined by the Rietveld method to reach
a reasonable degree of precision [15].
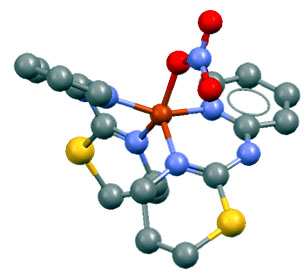
This resolution strategy has been successfully applied to the structural determination of different substances of biological interest in collaboration with the Synthesis Group of the Department of Inorganic Chemistry of the University of Extremadura. This group works mainly on the synthesis of coordination complexes (which, essentially, are formed by joining a metallic cation to an organic or organometallic anion called ligand), so that the main structures resolved belonged to these. The first one was the complex [Cu(NO)3-(PITz)2]NO3 (with PITz = 2-(2-pyridil)iminotetrahidro-1,3-thiazine) [15]; the second was the complex ZnCl2(TdTz) (with TdTz = 2-(3,4-dichlorephenil)imino-N-(2-thiazin-2-il)thiazolidine) [17]. The analysis of the bond distances and angles and torsion angles fulfilled all the chemical requirements, so that both solutions were considered satisfactory, and were successfully deposited in the CCD (Cambridge Crystallography Database) database.
.jpg) Recently, a new ab initio method for structural
resolution has received considerable attention. The nucleus of
this method consists of a relatively simple algorithm –the charge
flipping algorithm. This algorithm is a cyclic process, each cycle
containing four stages. Initially, random phases are assigned to
the structure factor amplitudes derived from the observed
intensities, which allows plot a (random) charge density map ρ(r).
In the first stage, the signs of all the electron densities in the
map below a certain threshold value δ (being δ
a small positive number) are reversed, which results in a modified
electron density map g(r). In
the second stage, a fast Fourier transform (FFT) allows a set {G(q)}
of temporary structure factors to be generated. In the third
stage, the phases of these G’s are combined with the
moduli of the original structure factors, which gives rise to a
second set {H(q)} of structure
factors, whose phases are no longer random. Finally, the inverse
FFT allows plot a new charge density map, which is used as
starting point for the second cycle. The iterative process goes on
until convergence is reached. The charge flipping method can solve
the structure in the space group P1, so that no previous
knowledge about symmetry is required. Information about either the
chemical composition or the type of atoms presents is not needed
either, so that the procedure is indeed ab initio. The
method has been successfully applied in two relatively complex
situations. In the first one, it was used to solve the structure
of [NBu4]2[Pd2{C4(COOMe)4}2(µ-OH)2], characterized by having a
relatively high number of atoms (156 non hydrogen ones) in its
asymmetric unit [29]. In the second one, the algorithm was used to
solve the structure of ZnCl2(BzTz)2 [with BzTz =
N-(5,6-dihydro-4H-1,3-thiazin-2-yl)-2-aminobenzimidazole], which
is characterized by exhibiting continuous positional disorder in
some of its atoms [30].
Recently, a new ab initio method for structural
resolution has received considerable attention. The nucleus of
this method consists of a relatively simple algorithm –the charge
flipping algorithm. This algorithm is a cyclic process, each cycle
containing four stages. Initially, random phases are assigned to
the structure factor amplitudes derived from the observed
intensities, which allows plot a (random) charge density map ρ(r).
In the first stage, the signs of all the electron densities in the
map below a certain threshold value δ (being δ
a small positive number) are reversed, which results in a modified
electron density map g(r). In
the second stage, a fast Fourier transform (FFT) allows a set {G(q)}
of temporary structure factors to be generated. In the third
stage, the phases of these G’s are combined with the
moduli of the original structure factors, which gives rise to a
second set {H(q)} of structure
factors, whose phases are no longer random. Finally, the inverse
FFT allows plot a new charge density map, which is used as
starting point for the second cycle. The iterative process goes on
until convergence is reached. The charge flipping method can solve
the structure in the space group P1, so that no previous
knowledge about symmetry is required. Information about either the
chemical composition or the type of atoms presents is not needed
either, so that the procedure is indeed ab initio. The
method has been successfully applied in two relatively complex
situations. In the first one, it was used to solve the structure
of [NBu4]2[Pd2{C4(COOMe)4}2(µ-OH)2], characterized by having a
relatively high number of atoms (156 non hydrogen ones) in its
asymmetric unit [29]. In the second one, the algorithm was used to
solve the structure of ZnCl2(BzTz)2 [with BzTz =
N-(5,6-dihydro-4H-1,3-thiazin-2-yl)-2-aminobenzimidazole], which
is characterized by exhibiting continuous positional disorder in
some of its atoms [30].
3. Mechanical properties of materials at intermediate temperatures
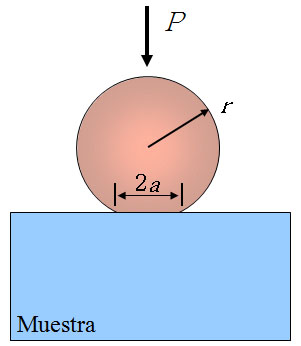 Ceramic materials are brittle, so that at temperatures below the
creep ones do not exhibit plasticity. The magnitudes of interest
are different to the relevant at high temperatures. For instance,
at room temperature, the study of these systems consists of
performing alternative mechanical tests, like those of
indentation, which allow determine magnitudes such as elastic
modulus, hardness, toughness or fracture strength. Among these
tests, the indentation with spheres (or Hertz indentations) are
particularly important because the contact is initially elastic.
By means of this type of tests one can get the indentation stress
- strain curve and, from it, information about the mechanical
properties of a material.
Ceramic materials are brittle, so that at temperatures below the
creep ones do not exhibit plasticity. The magnitudes of interest
are different to the relevant at high temperatures. For instance,
at room temperature, the study of these systems consists of
performing alternative mechanical tests, like those of
indentation, which allow determine magnitudes such as elastic
modulus, hardness, toughness or fracture strength. Among these
tests, the indentation with spheres (or Hertz indentations) are
particularly important because the contact is initially elastic.
By means of this type of tests one can get the indentation stress
- strain curve and, from it, information about the mechanical
properties of a material.
.jpg) Part
of our research has been useful to develop a new methodology to
extend the used at room temperature to temperatures close to creep
ones [22]. To that end, a high-temperature furnace was coupled to
the Hertz indentation device (built with the proper refractory
material, namely alumina or silicon nitride) to obtain the
indentation stress-strain curve at a given temperature. The
elastic modulus of the material at that temperature was trivially
calculated from the lineal part of the curve. Subsequently, the
stress-strain curve is modeled as the superposition of two
regions, one corresponding to a purely elastic contact and the
other to a plastic deformation with a given work hardening
parameter; the adjustable parameters are thus the yield stress (Y)
and the work hardening parameter (n). Using the finite element
method (FEM) these parameters are fitted by trial and error until
obtaining a simulated stress-strain curve which overlaps with the
experimental one; when it happens, the calculated Y and n
parameters are those of the material at the given temperature.
This method has been successfully applied to the mechanical
behavior at intermediate temperatures of polycrystalline Al2O3
[25] and ZrO2 [27].
Part
of our research has been useful to develop a new methodology to
extend the used at room temperature to temperatures close to creep
ones [22]. To that end, a high-temperature furnace was coupled to
the Hertz indentation device (built with the proper refractory
material, namely alumina or silicon nitride) to obtain the
indentation stress-strain curve at a given temperature. The
elastic modulus of the material at that temperature was trivially
calculated from the lineal part of the curve. Subsequently, the
stress-strain curve is modeled as the superposition of two
regions, one corresponding to a purely elastic contact and the
other to a plastic deformation with a given work hardening
parameter; the adjustable parameters are thus the yield stress (Y)
and the work hardening parameter (n). Using the finite element
method (FEM) these parameters are fitted by trial and error until
obtaining a simulated stress-strain curve which overlaps with the
experimental one; when it happens, the calculated Y and n
parameters are those of the material at the given temperature.
This method has been successfully applied to the mechanical
behavior at intermediate temperatures of polycrystalline Al2O3
[25] and ZrO2 [27].
In addition to these studies, within this research line I have collaborated in the study of the fatigue of silicon single crystals [26]. Due to the highly covalent nature of its bonding, silicon has traditionally been regarded as a material immune to slow crack growth (SCG) and, therefore, to mechanical fatigue. Let us consider then that silicon is submitted to Hertz indentation tests under some indentation load (well below the failure load but higher than the critical load for initiation of cone cracks) kept for different time intervals. One of the ways to measure the strength of silicon under these conditions consists of fixing it to a transparent and soft substrate (for instance, a polycarbonate), with the damage in contact with the substrate, and to apply a increasing load with a sphere until some damage is observed. Normally, this is a radial crack which finally causes failure. The load under which the radial crack reaches the silicon - polycarbonate interface allows calculate the fracture strength of silicon. If silicon is immune to SCG, then its strength should be the same regardless the time of application of the indentation load; this is indeed which one observes experimentally.
.jpg) Now, if the applied indentation load is not static, but cyclic
(that is, if the peak load is applied at a given frequency), then
a sharp decrease in the strength is observed at a critical number
of cycles, which demonstrates that silicon exhibits fatigue under
these conditions. This same effect has been observed under
different indentation loads. The implications in the behavior of
microelectromecanic and nanoelectromecanic systems are evident.
The original work [26] constituted the first experimental evidence
of fatigue in silicon single crystals. In addition, it was
possible to identify the fatigue mechanism [28], which shows three
clear stages:
Now, if the applied indentation load is not static, but cyclic
(that is, if the peak load is applied at a given frequency), then
a sharp decrease in the strength is observed at a critical number
of cycles, which demonstrates that silicon exhibits fatigue under
these conditions. This same effect has been observed under
different indentation loads. The implications in the behavior of
microelectromecanic and nanoelectromecanic systems are evident.
The original work [26] constituted the first experimental evidence
of fatigue in silicon single crystals. In addition, it was
possible to identify the fatigue mechanism [28], which shows three
clear stages:
4. Numerical simulation of solids at atomic scale
 During last years, simulation techniques at atomistic scale
(particularly Molecular Dynamics and its variants) have been
successfully applied due to the hardware and algorithm
development. The success of these lies essentially on its low
cost, in the possibility to accurately control microscopic
parameters and to achieve experimental conditions hard to find in
conventional laboratories. In particular, my interest focuses on
the study of the structure and dynamics of the grain boundaries in
a polycrystal. This study is a fundamental aspect in the
development of advanced materials due to the effect that these
boundaries have in their macroscopic behavior. The grain
boundaries are structural defects of relatively high energy which
constitute preferential regions for the localization of other
defects affecting the physical properties; in addition, the
movement of the grain boundaries controls kinetics during the
processing of polycrystals, and therefore determines morphological
parameters such as grain size or texture, on which those
properties depend. Anyway, the knowledge of the dynamics of grain
boundaries at atomistic scale is a first step when one tries to
develop a multiscale model for plasticity in polycrystals.
During last years, simulation techniques at atomistic scale
(particularly Molecular Dynamics and its variants) have been
successfully applied due to the hardware and algorithm
development. The success of these lies essentially on its low
cost, in the possibility to accurately control microscopic
parameters and to achieve experimental conditions hard to find in
conventional laboratories. In particular, my interest focuses on
the study of the structure and dynamics of the grain boundaries in
a polycrystal. This study is a fundamental aspect in the
development of advanced materials due to the effect that these
boundaries have in their macroscopic behavior. The grain
boundaries are structural defects of relatively high energy which
constitute preferential regions for the localization of other
defects affecting the physical properties; in addition, the
movement of the grain boundaries controls kinetics during the
processing of polycrystals, and therefore determines morphological
parameters such as grain size or texture, on which those
properties depend. Anyway, the knowledge of the dynamics of grain
boundaries at atomistic scale is a first step when one tries to
develop a multiscale model for plasticity in polycrystals.
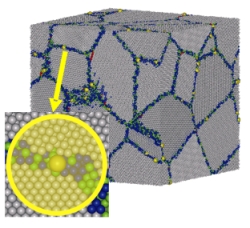 The dynamics of grain boundaries is extremely sensitive to the
presence of impurities. This fact is crucial in doped
polycrystals, where, under some conditions, the solute atoms tend
to segregate to the grain boundaries to reduce the energy of
lattice deformation; the importance of segregation lies on that it
couples to the dynamics of grain boundaries. There have been
developed several theoretical models for the impurities
segregation in grain boundaries, which have either an
thermodynamical/statistical nature or use a space-charge point of
view. Generally, these works provide with quantitative information
about segregation, although they neglect the atomistic structure
of the grain boundaries and the interactions between constituents
at that scale, which is a handicap. In addition, most of the
mentioned models deal with equilibrium conditions, quite fa away
from the real situation in working conditions. There are also
models for dynamics of grain boundaries in presence of impurities.
The dynamics of grain boundaries is extremely sensitive to the
presence of impurities. This fact is crucial in doped
polycrystals, where, under some conditions, the solute atoms tend
to segregate to the grain boundaries to reduce the energy of
lattice deformation; the importance of segregation lies on that it
couples to the dynamics of grain boundaries. There have been
developed several theoretical models for the impurities
segregation in grain boundaries, which have either an
thermodynamical/statistical nature or use a space-charge point of
view. Generally, these works provide with quantitative information
about segregation, although they neglect the atomistic structure
of the grain boundaries and the interactions between constituents
at that scale, which is a handicap. In addition, most of the
mentioned models deal with equilibrium conditions, quite fa away
from the real situation in working conditions. There are also
models for dynamics of grain boundaries in presence of impurities.
The challenge consists then of studying the dynamics of grain boundaries and its coupling to impurities segregation under non-equilibrium conditions by numerical simulation. A number of the works on simulation of dynamics of grain boundaries has been devoted to the study of their mobility as a function of the microstructural parameters or the external variables. By means of Molecular Dynamics (MD) it has been possible to show then that grain boundary migration and sliding occur simultaneously and, if the geometrical constraints imposed by the triple points of the grain structure are not severe, both give rise to rotation of the grains themselves. MD has also been successfully used to study Coble creep and the role played in it by grain boundary sliding and migration. Finally, it is also noteworthy that MD has allowed study the correlation between grain boundary sliding, diffusional creep and normal grain growth.
Despite the advance associated to the use of simulation techniques, there are many open problems, some of which are crucial. In most of the works, impurities are randomly distributed, and their evolution takes place either by pure diffusion or due to the action of a driving force which causes the movement of the grains. In no case the characteristics of the impurities diffusion or the interactions between them are studied. Regarding the movement of the grain boundaries, it is commonly regarded that they evolve under the action of temperature; the effect of external stresses has not been systematically studied. In addition, it is generally accepted that the movement of the grain boundaries is induced by a driving force which is proportional to the grain curvature. However, when aliovalent impurities are present, a local electric field at the grain boundaries exists, which affects diffusivity. The existence of this field has an important effect on the dynamics of grain boundaries, which causes, among others, the apparition of a threshold stress below which no sliding occurs. Finally, there is no satisfactory explanation for the plasticity of nanometric polycrystals, which cannot be extrapolated from the laws valid for micro- and submicrometric ones.
 LINKS
LINKSInstitute of
Advanced Scientific Computation (ICCAEx)
NIST (National Institute of Standards and Technology, Gaithersburg, MD, USA)
| Home · Teaching · Personal (in Spanish only) |
Juan J. Meléndez
|